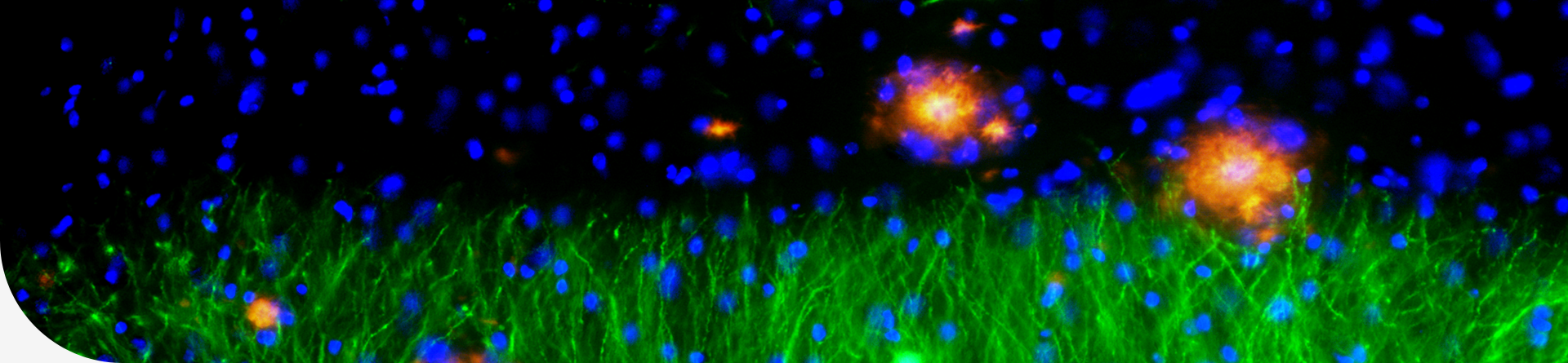
Myocardial hypertrophy, characterized by an increase in cardiac muscle mass, is an adaptive response to pathological stimuli such as hypertension and valve disease. While initially beneficial, sustained hypertrophy may lead to further disease progression and exacerbate heart failure, which is one of the most common and deadly medical conditions worldwide and remains incurable. Inhibition or reversal of cardiac hypertrophy and subsequent maladaptive myocardial remodeling is the main treatments for heart failure.
Cardiomyocyte hypertrophy can be triggered by diverse intrinsic and extrinsic stimuli including mechanical stress and hormones that are recognized by cardiomyocytes via a range of cell membrane receptors. A broad spectrum of GPCR antagonists have been used to treat heart failure, including those targeting β-adrenergic receptors, angiotensin II receptor, and aldosterone receptor. Owing to inadequate efficacy and adverse effects, only a few drugs for cardiovascular diseases, especially for heart failure, have indications for improving quality of life, physical function, or symptoms. Therefore, it is imperative to identify new therapeutic targets and strategies to fight against heart failure.

Professor ZHANG Yan’s research team from the School of Medicine at Zhejiang University, partnered with Professor ZHANG Yan’s research group from the School of Basic Medical Sciences at Peking University to conduct groundbreaking research. Together, they developed an effective apelin receptor modulator, when compared to the established APLNR agonists, exhibits superior therapeutic effects against cardiac hypertrophy and reduced adverse effects, opening up a new avenue for the targeted development of cardiovascular disease drugs.
Their findings were published in the journal Cell on March 1.

Fig. 1: Prof. ZHANG Yan from Zhejiang University (middle) and his colleagues in the lab
G protein-coupled receptors (GPCRs) are the largest family of membrane receptors that serve as primary targets of ~1/3 of currently marketed drugs. The apelin receptor (APLNR, also known as APJ), a prototypical Class A GPCR, activates both G protein and β-arrestin signaling pathways through the endogenous ligand apelin. This dual activation modulates various physiological and pathological processes. Especially in the cardiovascular system, APLNR activation facilitates vasodilation, positive inotropy, angiogenesis diuresis, and lower blood pressure. Moreover, it regulates cardiovascular disease by inhibiting myocardial fibrosis, reducing pathological myocardial hypertrophy, and offering resistance against heart failure and pulmonary arterial hypertension. APLNR is therefore regarded as a promising therapeutic target for cardiovascular disease. However, adverse effects through the β-arrestin pathway limit its pharmacological use.
It is this “duality” that has baffled scientists, as it entails both positive effects and negative side effects, significantly impacting the efficacy and safety of drugs. Many world-renowned pharmaceutical companies and research institutions are striving to develop safe and effective APLNR agonists, but so far, no precise precisely targeted drug molecules have received approval for the market.

Fig. 2: A cartoon model of agonists- or stretch-induced APLNR signaling profile and potential pharmacological effects.
Professor ZHANG Yan’s team at Zhejiang University has long been committed to delving into transmembrane signal transduction mechanisms and devising precise control strategies. Their work centers on the development and implementation of innovative methods utilizing cryo-electron microscopy for GPCR pharmacology research. This approach empowers them to intervene in diseases with precision through structure-based design, finely tuning GPCR functions for therapeutic purposes.
Simultaneously, Professor ZHANG Yan’s team at Peking University has steadfastly focused on the mechanisms of myocardial injury and its implications for cardiovascular disease. Their research endeavors have led to the identification of targeted disease prevention and treatment strategies, opening the door for the prevention and treatment of cardiovascular disease.
The first step in this study involved unraveling the enigma of how different signal spectrum agonists activate receptors to mediate downstream signaling pathways.
The research team has successfully resolved the high-resolution cryo-EM structure of the APLNR-Gi1 complex, activated by the endogenous balance agonist apelin and two biased G protein agonists, MM07 and CMF-019. They found that the APLNR-Gi1 complexes bound to these three agonists appeared remarkably similar, resembling “triplets,” with seemingly no discernible difference.
However, the research team persisted and, after multiple experiments, finally identified subtle differences among them. If the ligand were likened to a key and the receptor to a lock, the top end of apelin, or the key handle, would be relatively extended, while MM07’s key handle would be bent into a ring. This resulted in differences in the depth and specific position of their key main parts inserted into the receptor lock, with the critical sites inserted into two pockets defined as “twin hotspots,” M11 and F13, affecting biased signal transduction.

Fig. 3: Structural basis of signal bias in APLNR
Furthermore, they have also revealed the signal transduction mechanism from the ligand binding pocket to downstream effector protein binding pocket—wherein D752.50, located at the hub of a polar network, acted as a switch for biased signaling, causing a 0.1 nanometer structural displacement. It is this minuscule difference in the spatial position, equivalent to one millionth the diameter of a strand of hair, that triggered distinct conformational changes in downstream effector protein-binding pockets and ultimately determined whether G protein signaling or β-arrestin signaling would be initiated.

Fig. 4: Allosteric network allows ligands to favor either intracellular conformation
According to Professor ZHANG Yan at Zhejiang University, “By uncovering the molecular mechanism through which different biased signaling molecules recognize and activate APLNR, we have designed two exclusive G protein-biased agonists WN353 and WN561. These agonists effectively suppress β-arrestin activity while preserving G protein signaling capability.”
To minimize interference, the team blended the newly designed agonists with existing ones on the market, scrambled their numbering, and utilized both in vitro cultured cardiomyocytes and in vivo animal models of heart disease for “double-blind” functional screening and validation. Dr. WANG Weiwei said, “I was extremely nervous when ‘the mystery box’ was opened. However, the results were reassuring, demonstrating that our G protein-biased agonists WN353 and WN561 did not incur any myocardial hypertrophy. Our experiments also reaffirmed the β-arrestin signaling pathway of APLNR as the primary pathway leading to myocardial hypertrophy.”
Subsequently, the researchers simulated the therapeutic effects of APLNR agonists in the context of myocardial hypertrophy, excluding pathological stimuli, such as valve disease or hypertension. The results revealed that Apelin exacerbated myocardial hypertrophy, while MM07 and CMF-019 had no effect. Encouragingly, the newly developed agonist WN561 demonstrated efficacy in alleviating myocardial hypertrophy in mice, regardless of pathological or non-pathological conditions, further showing the promising prospects of this novel G protein-biased APLNR agonist for the treatment of myocardial hypertrophy and heart failure.

Fig. 5: Graphical abstract
This work revealed the distinctive recognition properties of APLNR complexes when bound to various biased ligands. It also led to the rational design of APLNR agonists with absolute G protein signal selectivity. Through experimentation on three different animal models, the safety and efficacy of these newly devised active molecules were demonstrated for the development of cardiovascular medications targeting APLNR.

Myocardial hypertrophy, characterized by an increase in cardiac muscle mass, is an adaptive response to pathological stimuli such as hypertension and valve disease. While initially beneficial, sustain...

Tuesday, the Second Affiliated Hospital, Zhejiang University School of Medicine (SAHZU) and National Heart Centre Singapore (NHCS) signed a memorandum of understanding (MOU) to collaborate on clinical...
Excessive or repetitive fear play a significant role in the development of anxiety disorders, with the amygdala serving as the central locus for fear processing. Clinical research has demonstrated tha...